")
")
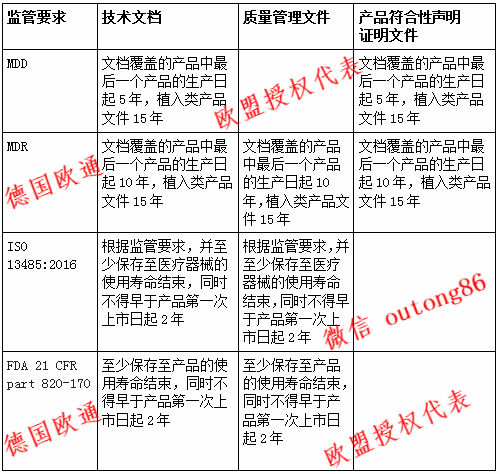
欧洲和美国的医疗法规对于文档保存期限有所不同,欧洲新规MDR已经颁布,而MDD仍然有效,德国欧通欧盟授权代表对MDR/MDD/FDA/13485法规里面关于文档保存期限的规定进行解读与对比。
另外,此前德国欧通已经为您介绍了关于技术文档的相关问答:不得不看!向欧代提交技术文件的10个知识问答
对欧盟授权代表或FSC自由销售证书感兴趣,欢迎添加微信outong86

-
-
技术文档(例如产品规格,临床评估,风险管理文件,操作性能记录)
-
产品符合性声明,公告机构出具的证书
-
质量管理体系文件,特别是规范文件和记录(例如流程和程序说明)
2保存期限:法规要求
MDD在附录7第2节关于产品符合性声明有如下规定:
“生产商将第3节中指定的技术文档编辑成册。 生产商或其授权代表应将这些文档以及产品符合性声明从该产品停产日起保存至少5年,以应对供所属国家主管部门检查。 对于植入类产品,这个保存期限为产品停产日起至少15年。”
技术文档、产品符合性声明的规定
MDR中的第10条(“生产商的一般义务”)规定非植入类产品的保存期限与MDD相比增加了一倍:
“(8) 生产商必须将技术文档,欧盟产品符合性声明以及根据第56条规定出具的所有证明包括任何修改和增补的副本,自欧盟产品符合性声明涵盖的最后一个产品投放欧盟市场起至少保存10年,以在需要时可以随时提供给主管当局。植入类产品文档的保存期为自文档涵盖的最后一个产品投放欧盟市场之日起15年。”
进口商也必须遵守有关产品符合性声明和所有相关证书的保存期限的规定,即自产品营销结束日起10年。
如果属于一个系统或一套医疗器械的不同产品适用于不同的保存期限规定的话,则以保存期限最长的规定为准(第22条)。
质量体系管理文件
附录9介绍了质量管理体系及其文件的要求。 其中第三章与行政规定有关的内容做出了关于保存期限的规定:
“生产商或者其欧盟授权代表(如果生产商在欧盟成员国没有注册分公司的话)必须把以下文件自文件涵盖的最后一个产品投入欧盟市场日起至少保存10年,对于植入类产品则为15年,以根据需要提交给相关的主管当局:
•欧盟产品符合性声明,
•第2.1节中第五个横线后提到的文档,尤其是第2.2节第2款c点中提到的数据和记录;
•第2.4节中有关更改的信息,
•第4.2节中列举的文件
•本附件提及的公告机构的决定和报告。”
以上第二点中引用的文件是指“生产商质量管理体系文件”,包括所有记录和过程或过程描述:
•验证,证明
•合格评定
•基本安全和性能要求的确定和证明,包括相应的“通用规格”证明
•风险管理
•临床评估,上市后临床随访
•产品唯一标识(UDI)
•各个生产阶段的规格
•“表述或质量管理体系变化的处理”
总结:简而言之,技术文档,所有基本规范文件(例如标准操作程序),以及质量管理体系的记录的保存期限为10年。
FDA在21 CFR 820.170部分中规定了对保存期限的要求。主管部门规定:
“(b)记录保存期限。 所有在本章中提及的文件必须至少被保存至产品寿命结束为止,同时不得早于自生产商上市销售之日起2年。”
ISO 13485:2016的规定并不像MDD或MDR那样具体。 它只要求相关单位必须设置保存期限。这个期限的设置必须能够保证“所有的技术文档至少在该产品的使用寿命期限之内得以保存”。在设定保存期限时,生产商必须确保文档的保存期限
-
不得短于“所有记录的保存期限”,而该期限不得短于“医疗器械通过入市审批后的2年”
-
不得短于监管部门的要求。
可见,MDR对技术文档保存期限的要求通常高于ISO 13485:2016的要求。
MDR/MDD/FDA/13485对文档保存期限对比